활용사례
활용사례는 주피터 노트북 형식의 파일(.ipynb)을 다운로드할 수 있는 형태로 제공하고 있습니다. 약 14개의 파일을 다운받을 수 있으며, 해당 문서를 참고하여 다른 종류의 오믹스 데이터 분석 또한 가능합니다.
활용사례는 어떻게 제공되나요?
헬∙빅∙쇼 사업의 데이터 활용 사례는 실행가능한 프로그램형 문서인 Jupyter notebook으로 제공합니다. 본 문서를 통해 직접 분석 코드를 실행하거나, 원하는 형태로 변경하여 분석할 수 있습니다. 또한, “전처리 파이프라인” 메뉴에서 제공한 결과가 본 Jupyter notebook의 입력 자료로 활용됩니다. Python 3.10 을 기준으로 작성하였으며, 주피터 노트북(.ipynb) 형식으로 저장하여 제공합니다.
본 문서를 참고하여 다른 종류의 오믹스 데이터 분석을 수행할 수도 있으며 잘못된 부분이나, 개선이 필요한 코드는 dsc@insilicogen.com 으로 알려주세요. 지속적으로 피드백 반영 예정입니다.
활용사례 문서
다운로드 준비중
종류
설명
질병별 오믹스 특징 (정상군)
모든 오믹스 자료에 대해 정상과 타 그룹간 차이 분석
질병별 오믹스 특징 (고지혈증군)
모든 오믹스 자료에 대해 고지혈증과 타 그룹간 차이 분석
질병별 오믹스 특징 (유방암군)
모든 오믹스 자료에 대해 유방암과 타 그룹간 차이 분석
질병별 오믹스 특징 (대장암군)
모든 오믹스 자료에 대해 대장암과 타 그룹간 차이 분석
질병별 오믹스 특징 (위암군)
모든 오믹스 자료에 대해 위암과 타 그룹간 차이 분석
참여자별 오믹스 특징
참여자 ID를 입력하면 모든 오믹스 자료 분석 결과 표시
유전자별 오믹스 특징
유전자 ID를 입력하면 모든 오믹스 자료 분석 결과 표시
WGS 변이 시그니처 분석
변이 종류별 카운팅 자료를 NMF 특징 추출하고 시그니처로 확인
WGS vs. Liquid biopsy 차이 분석
WGS, Liquid biopsy 동일 샘플에 대해 어떤 차이점이 있는지 확인
질병별 오믹스 특징 (고지혈증군)
모든 오믹스 자료에 대해 고지혈증과 타 그룹간 차이 분석
WGS vs. RNA-seq eQTL 연구
특정 유전변이가 특정 유전자발현에 영향을 미치는지 eQTL 연구
RNA-seq vs. Methyl-seq 연관 연구
유전자 수준 전사체 - 후성유전체 연관 연구
마이크로바이옴 다양성과 FB ratio 연구
참여자별 마이크로바이옴 다양성과 타 표현형 연관여부 확인 연구
멀티오믹스 딥러닝 연관 연구
WGS, RNA-seq, Methyl-seq 멀티오믹스 딥러닝 연관 연구
멀티오믹스 딥러닝 나이 추정 연구
WGS, RNA-seq, Methyl-seq 멀티오믹스 딥러닝 나이 추정 연구
활용사례 다운로드는 PC 버전에서만 가능합니다.
전사체 데이터 (RNA 데이터) 위암/대장암/정상군 비교 분석
목적 및 배경
기존 연구에서는 위암, 유방암에서의 변이 연관성 분석 및 GWAS 분석이 많이 발생 하였지만 암생존자의 body mass index (BMI) 정보를 이용하여 유전적 특성 및 변이를 탐색하는 연구 결과는 많이 보고 되지 않음.
- 이번 연구는 암생존자와 건강한 대조군 사이에서의 GWAS 분석을 통한 암생존자와 BMI 관련 유의한 유전자 변이 발견 및 연관성 확인
활용 데이터
암생존자와 건강한 군 100명
- 데이터 종류: WGS 데이터
분석 Tool
- Plink1.9, Rstudio, Bcftools, Tabix
연구 내용
- 환자별 WGS 데이터를 tabix 및 Bcftools를 이용하여 indexing 및 Merging을 진행 함.
-
Plink를 이용하여 합쳐진 WGS 데이터를 Binary 파일로 변환
* Phenotype의 경우 환자 별 BMI 정보를 이용 하여 진행 함.
-
GWAS 분석의 경우 Plink를 이용하여 BMI 와 암생존자와의 연관성 분석을 linear regression을 이용 하여 진행 함.
* GWAS 결과의 경우 유의하게 나온 table 정리 및 Rstudio를 이용하여 manhattan plot 생성 및 visualization
분석 결과
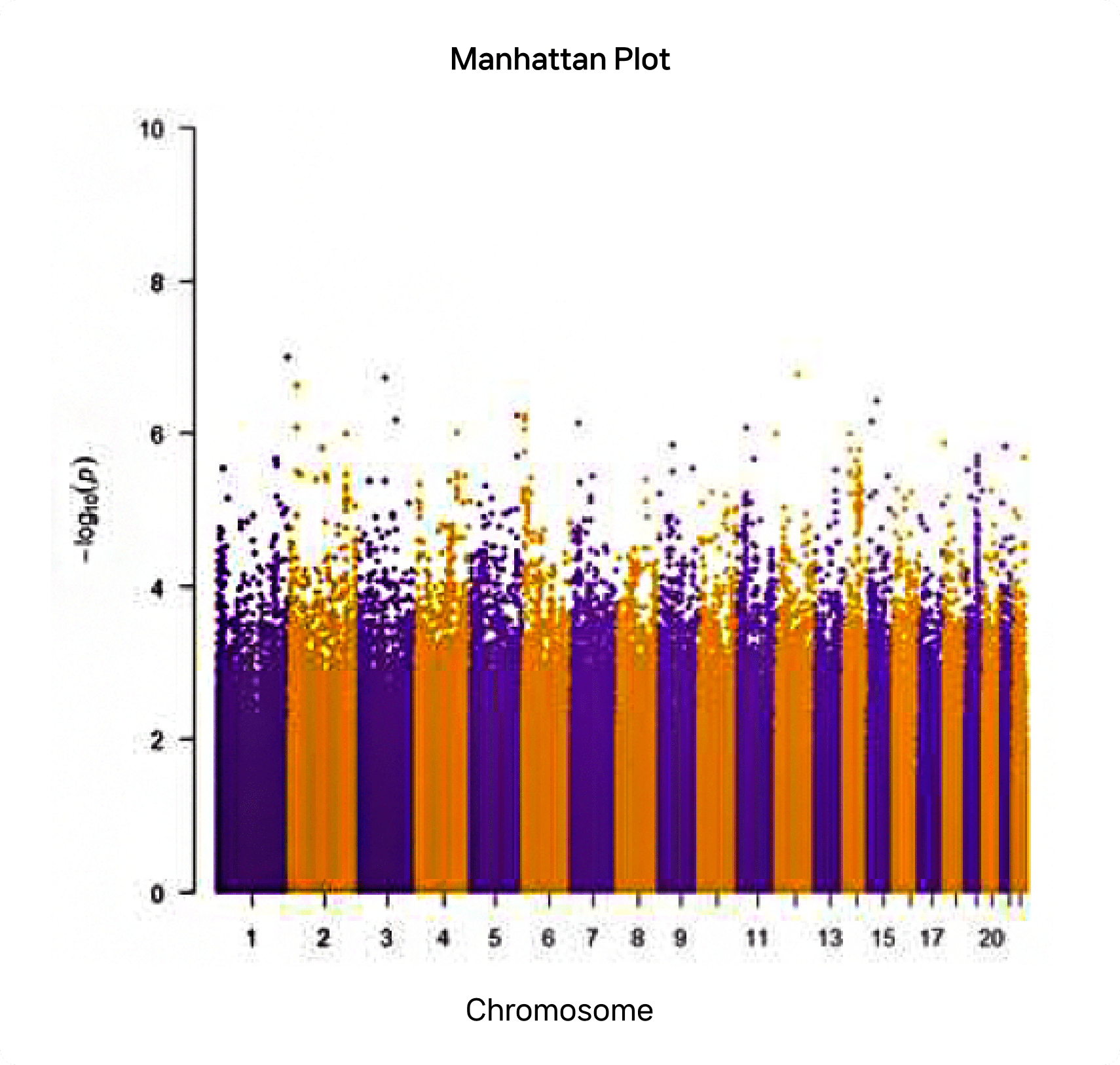
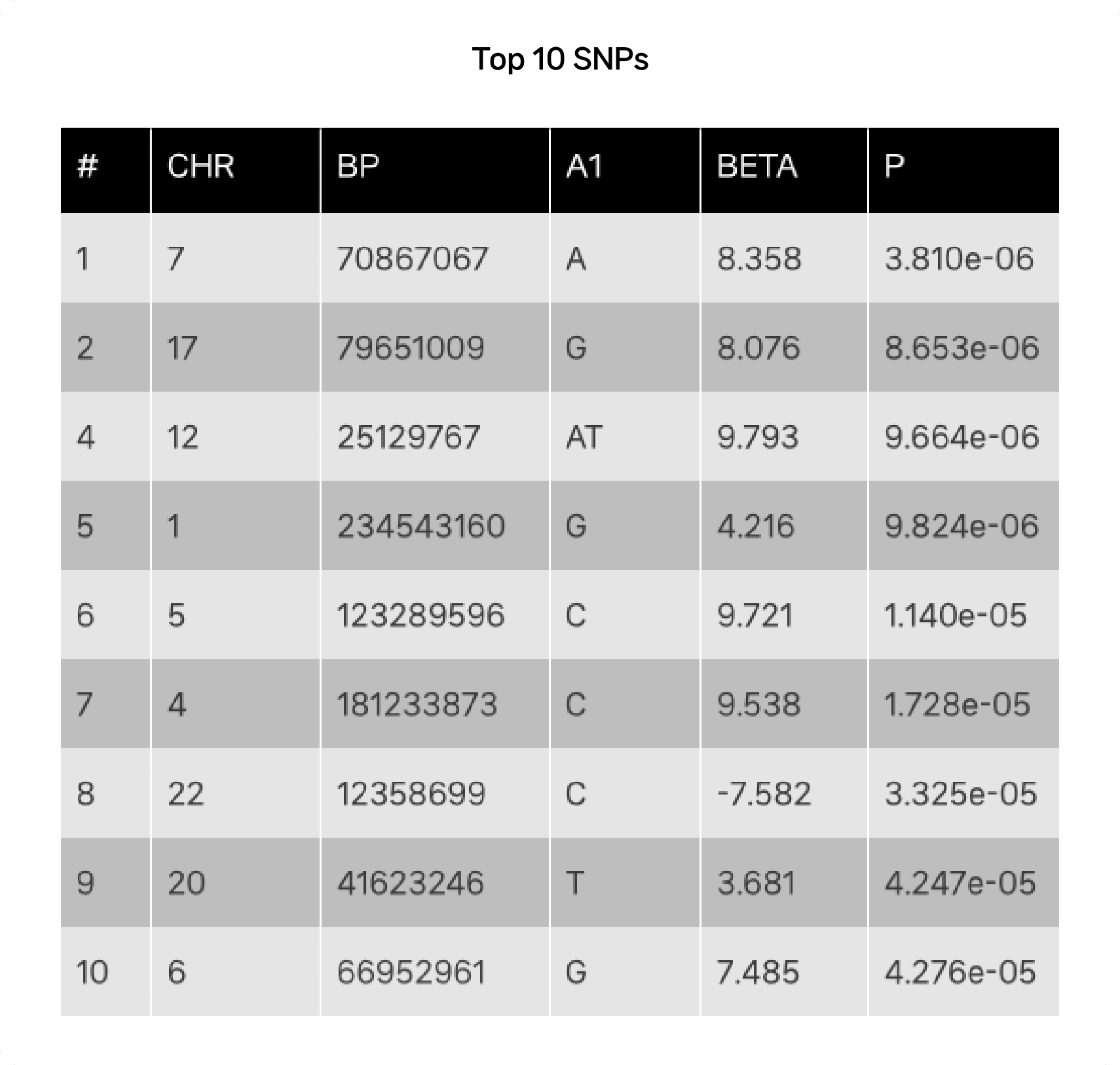
성과 및 활용 방안
- 해당 결과를 통해 기존 array 기반 GWAS에서 벗어난 WGS 데이터를 이용하여 보다 정확한 GWAS 분석이 가능해짐.
- 기존 array 기반에서 사용 되는 missing variant들을 추정하는 imputation 과정을 생략하고 실제 variant들을 이용한 정확한 분석이 가능함. 또한 암생존자와 BMI와의 연관성을 입증 할 수 있는 연구 성과. 해당 연구를 통해 암생존자의 비만 치료나 관련 연구에 이용 가능
Methyl-Seq 데이터를 활용한 후성유전학적 패턴 분석
목적 및 배경
일반인과 암생존자 사이의 DMR(Differentially methylatedregions) 분석 및 기능 분석
- 연령대와 성별에 따라 선택된 샘플들을 Control 및 Case 그룹으로 분류하고, DMR을 비교하여 유의미한 DMR 영역을 선별함.
- DMR과 근위에 위치한 유전자들의 GO 분석을 통해, hyper/hypo methylated 영역의 기능을 분석하여 정상인과 질환군 대비 methylation 차이가 실제로 어떤 기능을 regulation 하는지를 파악함.
활용 데이터
- 헬스케어쇼케이스 빅데이터 분석센터에 기구축된 일반인 194명 (건강인: 99 명, 만성질환자: 95 명)
- 암생존자 201 명 (유방암: 81 명, 위암: 39 명, 대장암:81 명)의 혈액 DNA로부터 생산된 Methyl-Seq 데이터
분석 Tool
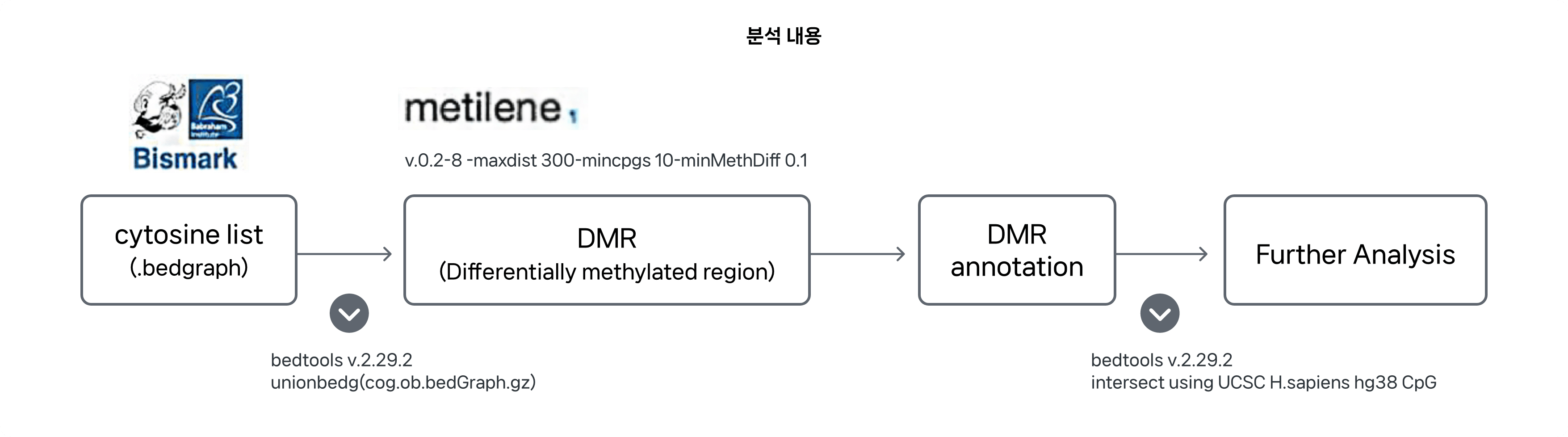
- bedtools v2.29.2: bedgragh(cytosine list)유틸, metilene v0.2.8: DMR 통계 계산 프로그램, DAVID: Gene enrichment 분석 웹 툴
연구 내용
- bedtools unionbedg를 사용하여 일반인, 암생존자 그룹을 각 각 g1, g2로 구분하여 한 파일로 통합
- metilene 툴을 사용하여 g1, g2 사이의 DMR을 계산. 이때 한 DMR 내 두 CpG 사이의 최대 거리는 300 nt, 최소 CpGs 10, 두 군간 평균 Methylation level 차이는 최소 0.1로 설정하여 DMR을 정의
- bedtools intersect를 사용하여 해당 DMR 영역 내, genomic context (gene/trnascript, promoter, CpG island, Sshore, Sshelf 등) UCSC H. sapiens hg38 CpG annotation 정보를 바탕으로 추가
- adjusted p-value(Mann-Whinney U-test) 0.05 미만, Methylation level 5 이상 시 “Hyper-methylation”, Methylation level -5 이하 시 “Hypo-methylation”인 유의미한 DMR 영역으로 구분
- 각 영역 내 포함하는 앙상블 transcripts ID로부터 DAVID GO Functional analysis를 진행
분석 결과
-
DMR distribution
* Metilene 결과 총 71,062 개의 영역에서 일반인과 암생존자의 패턴차이를 보였으며 이를 R base v.4.2.0을 사용하여 volcano plot 으로 시각화 함.
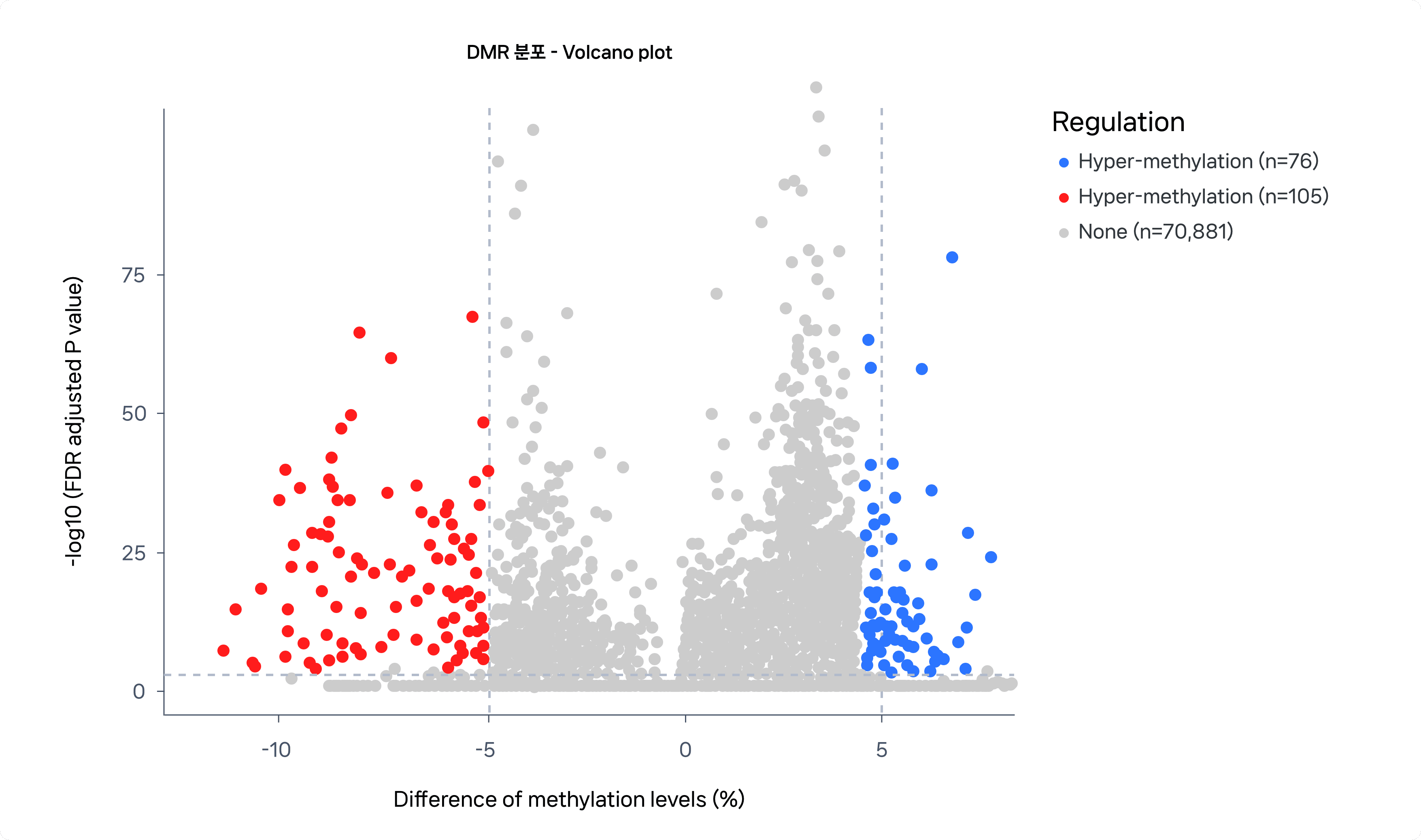
- 그 중 일반인군과 암생존자군에서 유의미한 패턴차이를 보인 Hyper-methylation 영역은 76개, Hypo-methylation 영역은 105개

-
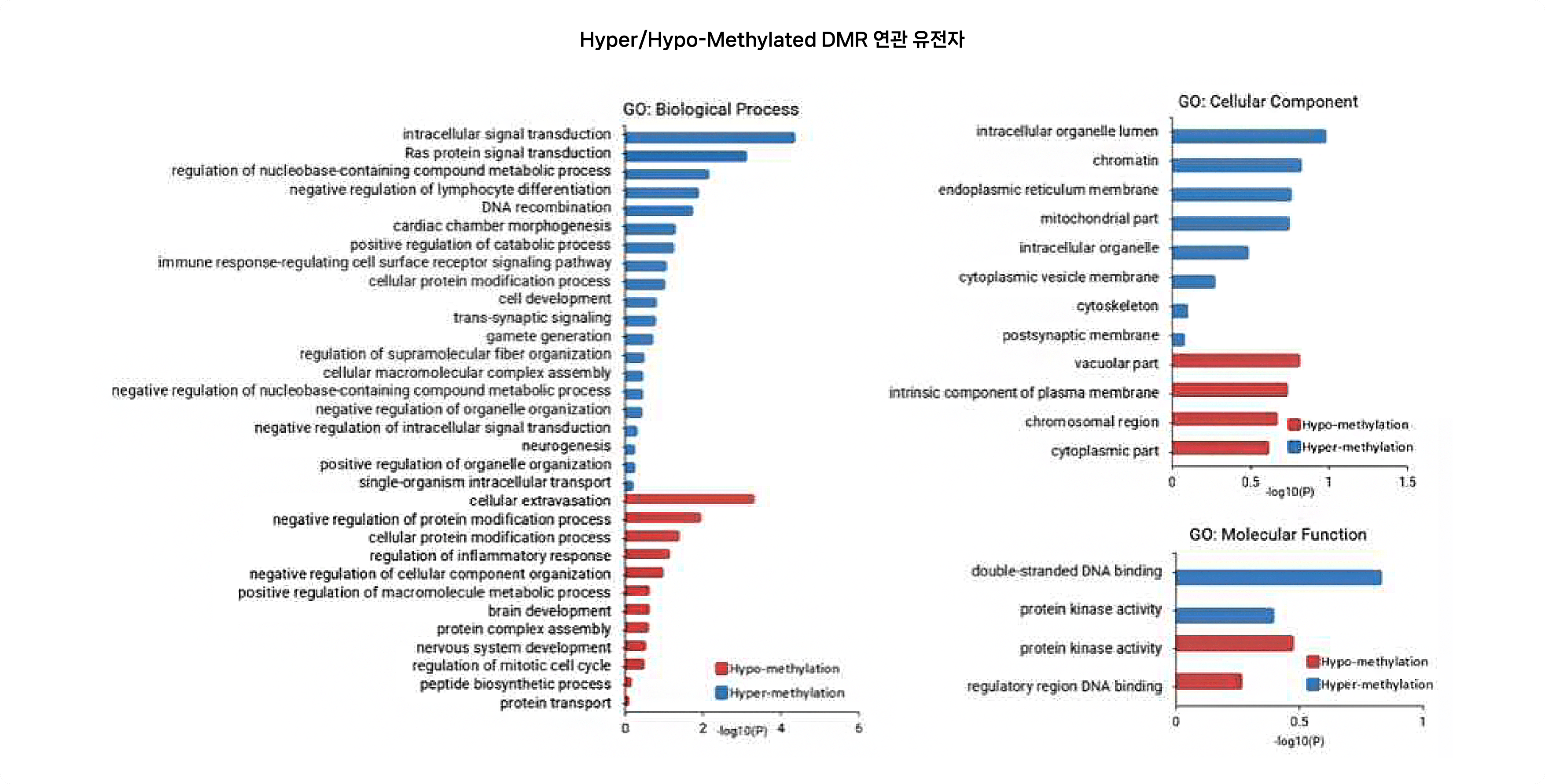
GO Function study for DMR related genes
* 각 유의미한 Hyper/Hypo-methylation 영역에 해당하는 transcripts의 수는 358개, 393개 였으며, 이를 DAVID Web Tool에서 Gene Ontology (GO)의 Biological Process, Cellular Component, Molecular Function 도메인에대해 맵핑을 진행하여 분석함.

성과 및 활용 방안
- 395명의 건강인, 만성질환자, 암생존자(유방암, 대장암, 위암)의 정도관리 된 샘플을 수집하고 데이터를 생산 및 표준화하여, 일반인과 암생존자 사이에서 발견된 유의적인 과메틸화, 저메틸화 영역 내 유전자의 GO 기능 분석을 수행한 첫 사례로, 일반인과 암생존자 공통 비교가 아닌 특정 암종의 세부적 분석도 가능할 것
Microbiome 데이터를 활용한 유의한 미생물 분석 사례 + 대사체 데이터 연관 분석
목적 및 배경
일반인과 암 생존자 사이에서의 유의미한 마이크로바이옴 선별 및 대사체 연관 관계 분석 실시
- 생산된 Metagenome Data를 기반으로 전체 균총의 서열 분석 진행
- Taxonomy classification & Diversity analysis를 통해 질환별, 정상군과의 차이를 보이는 균총을 분석
활용 데이터
- 일반인 136개 샘플, 만성질환자 60개, 유방암 생존자 61개, 대장암 생존자 52개, 위암 생존자 22개 총 331개의 마이크로바이옴 샘플
분석 Tool
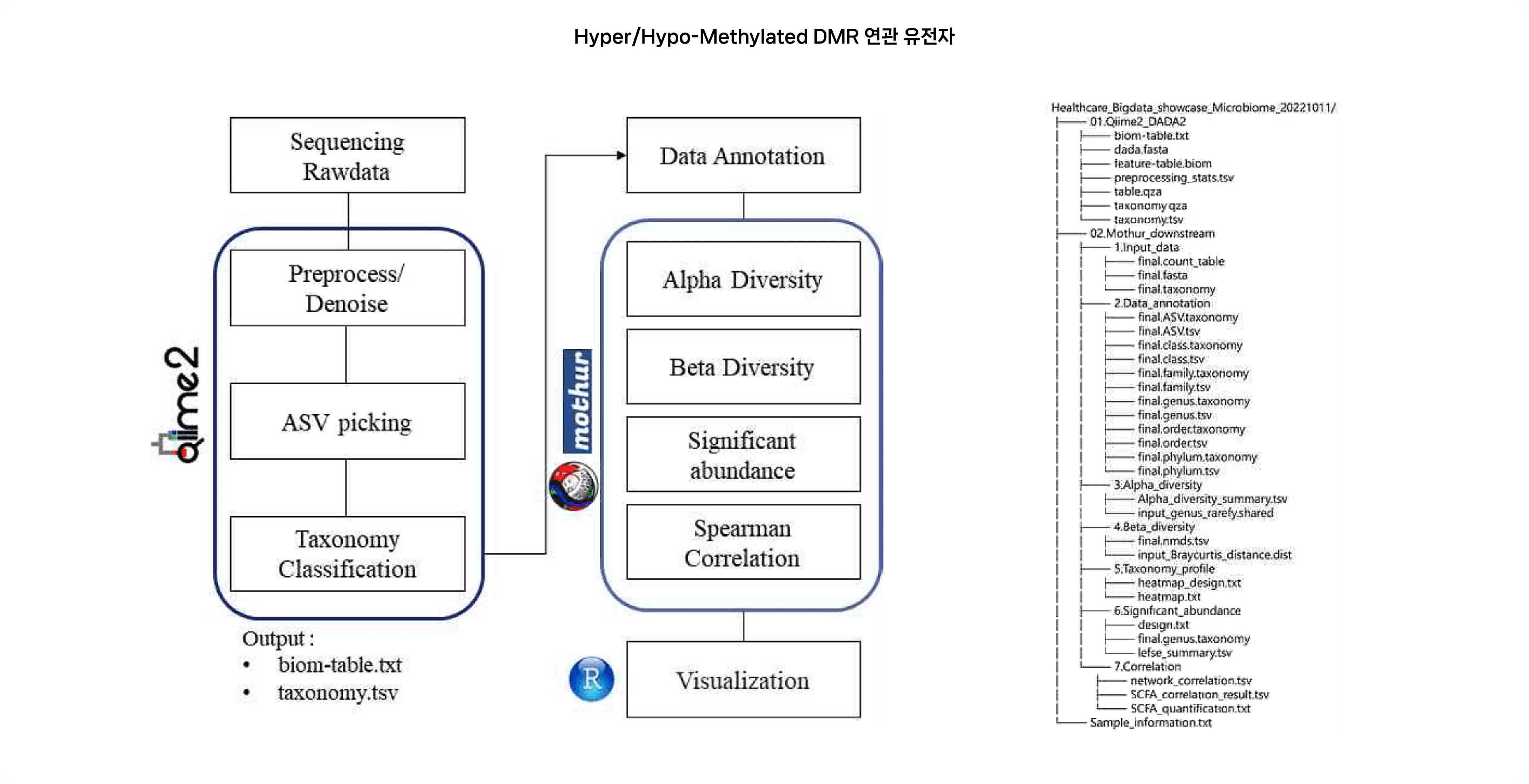
- Microbiome ASV 분석: QIIME2-DADA2, Downstream 분석: Mothur, Visualizaion: Rstudio, Cytoscape
연구 내용
- Illumina사의 Miseq Sequencing platform을 이용하여 16S rRNA V3-V4 region을 target으로 하여 301 paired-end Amplicon Sequencing 실시
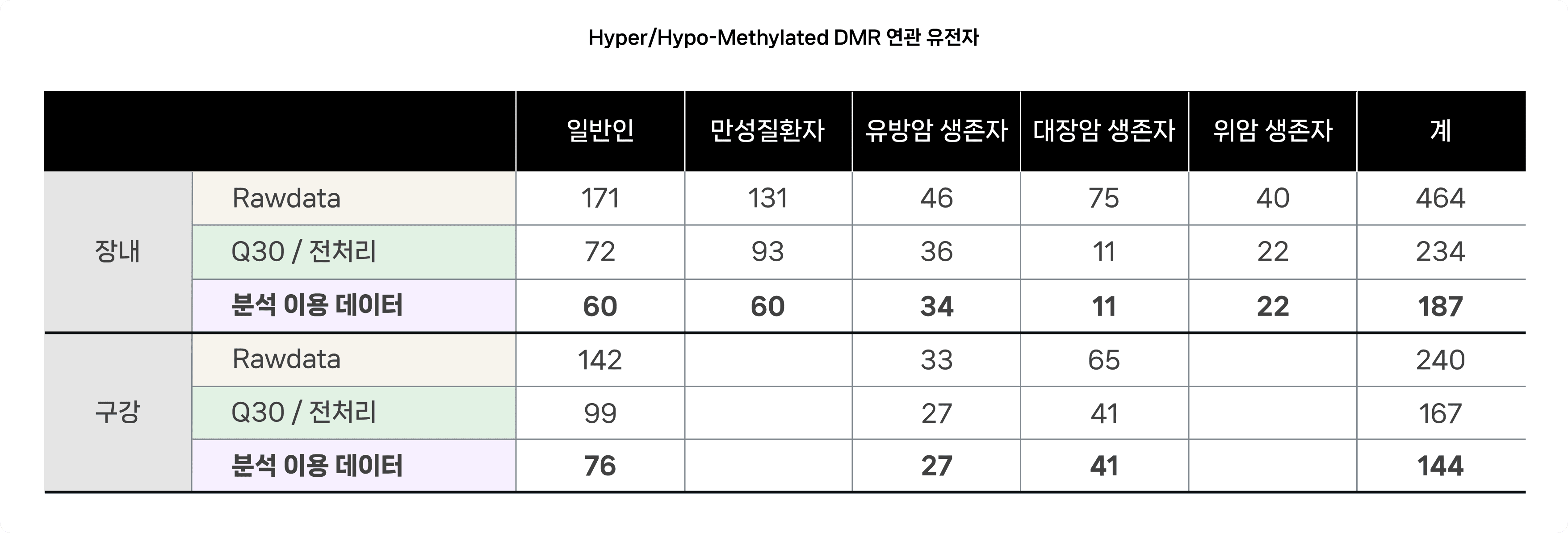
-
분석 전처리 과정 (최종 분석 이용 데이터 총 331개, gut: 187 / oral: 144)
* Q30 >= 65% AND 생산량 (10000 >= Reads) 기준을 충족한 샘플
* 환자 중복을 제거 (다수 채취 샘플의 경우 1회만 이용함.)
* Metabolites 정보가 있는 샘플 (*Metabolites file label 정보와 Sample정보가 일치하지 않아 환자 Label로 구분함.)
-
생산된 Reads를 Qiime2-DADA2 Software를 이용하여 Filtering 및 Denoise 과정을 거치면서 Chimera를 제거
* Chimera가 제거된 이후 ASV를 선발하였고, Silva Database 이용하여 Taxonomy classification 실시
* 샘플 간 거리를 나타내는 Beta diversity는 Bray curtis method를 이용하여 계산하였고, NMDS 분석을 실시
* R package vegan, ggplot2 등을 이용해 시각화
-
Downstream 분석은 Mothur Software를 활용
* 모든 샘플은 10,000 reads 로 rarefy 실시한 이후에 분석에 이용
* 샘플내의 다양성을 나타내는 Alpha diversity는 Richness의 지표인 Chao1과 Evenness도 포함하는 지표인 Shannon 두 가지에 대한 분석을 실시하였고, 그 결과는 R package ggplot2 등을 이용하여 시각화
-
그룹간 유의미한 Abundance의 미생물을 선별하는 선형판별분석인 LEfse Analysis도 실시
* LDA Score 3 이상의 Feature들만 선발하였고, R package ggplot2 등을 이용하여 시각화
* 상관관계 분석인 Spearman Correlation은 먼저 그룹간의 마이크로바이옴의 Network 분석을 위해 실시하였고, 그 이후에는 전체 샘플을 대상으로 SCFA 생산량 데이터와 상관관계 분석을 실시(P-value 0.05 이하의 feature들만 선별하여 시각화 실행)
분석 결과
-
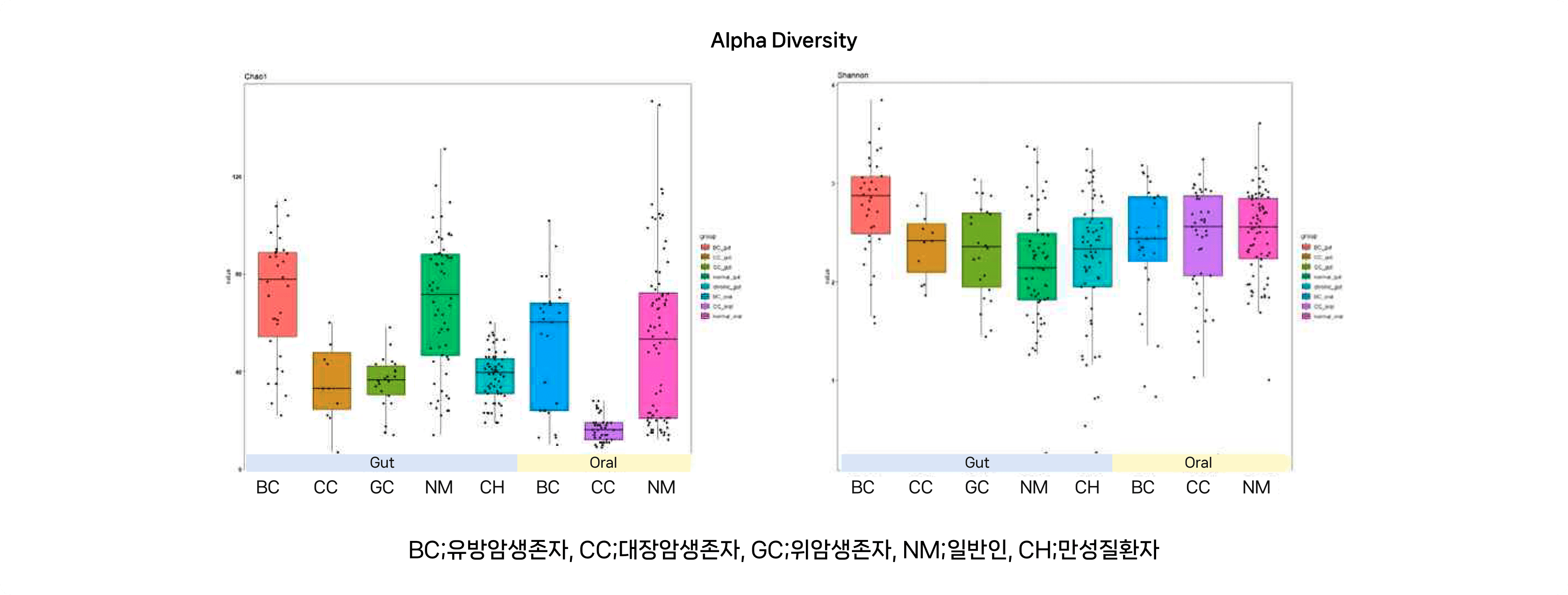
Alpha diversity
* Chao1의 결과를 볼때, 장내미생물에서는 대장암과 위암생존자, 만성질환자의 그룹에서 종 풍부도가 떨어지는 것을 볼 수 있고, 구강샘플의 경우 대장암 생존자 그룹에서 종 풍부도가 떨어짐을 확인 할 수 있다. 하지만 균등도까지 포함하는 Shannon의 결과에서는 비슷한 경향의 결과를 보임.
-
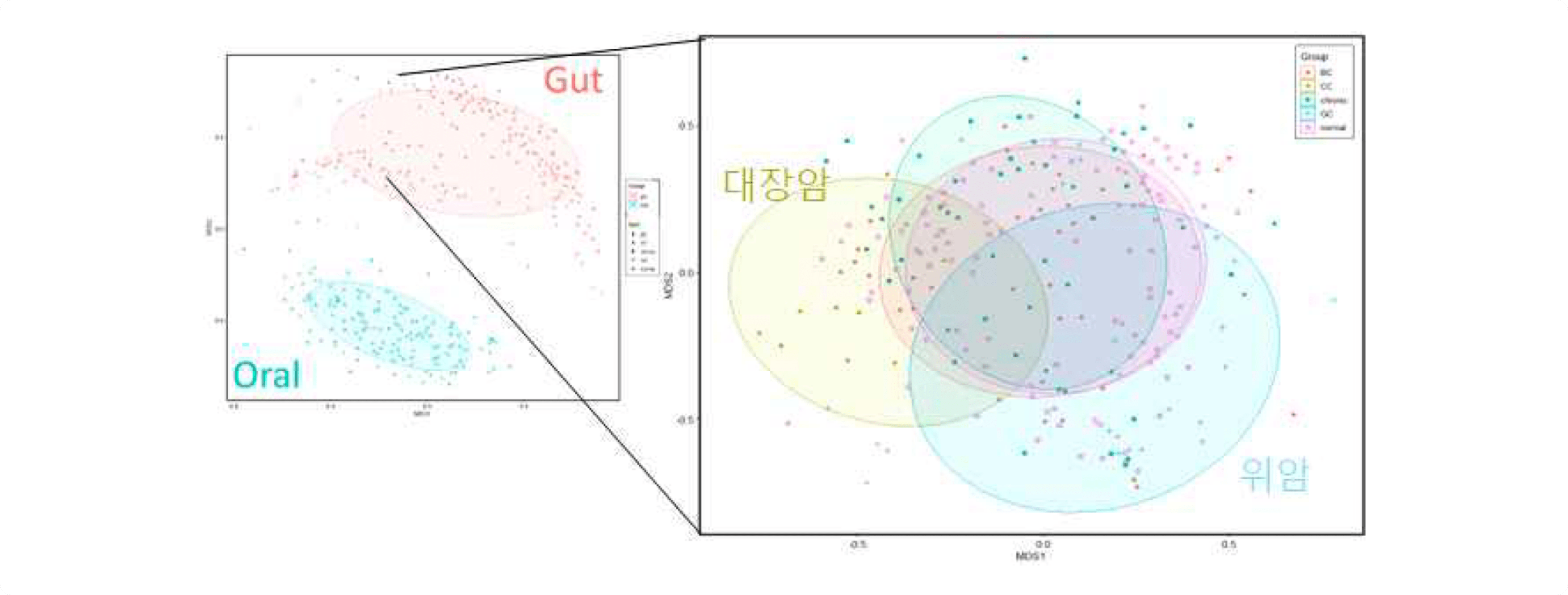
Beta Diversity
* 전체 샘플의 결과를 볼 때 Oral 샘플과 Stool 샘플의 차이가 극명하게 관찰 되며, 장내미생물 그룹내에서는 대장암과 위암그룹이 다른 그룹에 비해 유의미한 차이가 관찰됨.
-
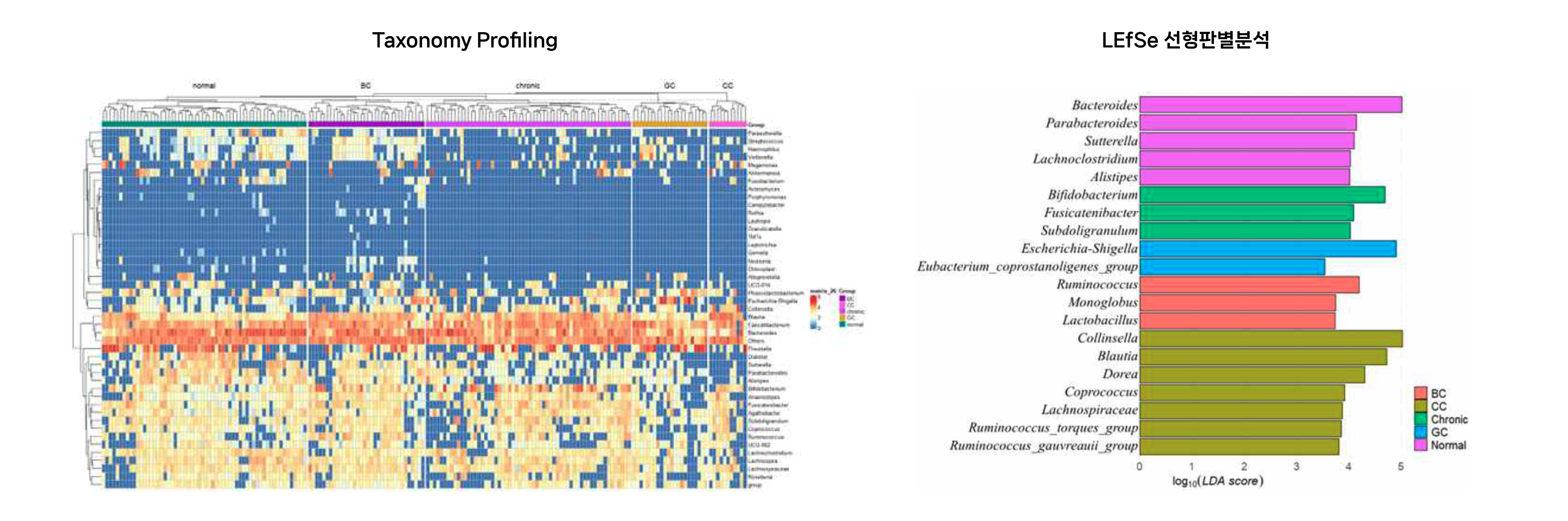
Significant Abundance Taxon
* 다른 암생존자 그룹과 비교했을때, 만성질환자와 일반인 그룹에서는 SCFA을 생산하는 유익균으로 잘 알려진 Bifidobacteria, Lactobacillus가 각각 검출
* 대장암 생존자 그룹에서는 Collinsella가 검출되었는데, 이는 실제로 대장암 환자군에서 더 많이 존재하는 균으로 보고 되고 있음 (PMID: 31611994)
성과 및 활용 방안
- 일반인과 암 생존자의 마이크로바이옴 비교 분석을 통해, 각 그룹간의 Alpha diversity 중 Choa1 의 유의미한 차이를 관찰 할 수 있음.
-
Beta diversity결과를 통해 구강 미생물 생태와 장내미생물 생태의 뚜렷한 차이를 관찰 하였으며, 또한 각 그룹간의 유의미하게 차이나는 마커 미생물을 선발할 수 있음.
* 특히 장내미생물과 밀접하게 관련있는 대장암 생존자 그룹에서 학계에 보고된 연구결과와 일치하는 분석 결과를 확인함.
Microbiome 데이터를 활용한 유의한 미생물 대사체 데이터 연관 분석
목적 및 배경
일반인과 암 생존자 사이에서의 유의미한 마이크로바이옴 선별 및 대사체 연관 관계 분석 실시
- 질병 (암, 만성질환)의 결과 혹은 원인에 따라 신체 대사의 활동의 부산물인 소변에서의 대사물은 차이가 있음.
활용 데이터
- 일반인 136개 샘플, 만성질환자 60개, 유방암 생존자 61개, 대장암 생존자 52개, 위암 생존자 22개 총 331개의 마이크로바이옴 샘플, 대사체 생산량(Acetate, Propionate, Butyrate) 데이터
분석 Tool
- Microbiome ASV 분석: QIIME2-DADA2, Downstream 분석: Mothur, Visualizaion: Rstudio, Cytoscape
연구 내용
- Illumina사의 Miseq Sequencing platform을 이용하여 16S rRNA V3-V4 region을 target으로 하여 301 paired-end Amplicon Sequencing 실시

-
분석 전처리 과정 (최종 분석 이용 데이터 총 331개, gut: 187 / oral: 144)
* Q30 >= 65% AND 생산량 (10000 >= Reads) 기준을 충족한 샘플
* 환자 중복을 제거 (다수 채취 샘플의 경우 1회만 이용함.)
* Metabolites 정보가 있는 샘플 (*Metabolites file label 정보와 Sample정보가 일치하지 않아 환자 Label로 구분함.)
-
생산된 Reads를 Qiime2-DADA2 Software를 이용하여 Filtering 및 Denoise 과정을 거치면서 Chimera를 제거
* Chimera가 제거된 이후 ASV를 선발하였고, Silva Database 이용하여 Taxonomy classification 실시
* 샘플 간 거리를 나타내는 Beta diversity는 Bray curtis method를 이용하여 계산하였고, NMDS 분석을 실시
* R package vegan, ggplot2 등을 이용해 시각화
-
Downstream 분석은 Mothur Software를 활용
* 모든 샘플은 10,000 reads 로 rarefy 실시한 이후에 분석에 이용
* 샘플내의 다양성을 나타내는 Alpha diversity는 Richness의 지표인 Chao1과 Evenness도 포함하는 지표인 Shannon 두 가지에 대한 분석을 실시하였고, 그 결과는 R package ggplot2 등을 이용하여 시각화
-
그룹간 유의미한 Abundance의 미생물을 선별하는 선형판별분석인 LEfse Analysis도 실시
* LDA Score 3 이상의 Feature들만 선발하였고, R package ggplot2 등을 이용하여 시각화
* 상관관계 분석인 Spearman Correlation은 먼저 그룹간의 마이크로바이옴의 Network 분석을 위해 실시하였고, 그 이후에는 전체 샘플을 대상으로 SCFA 생산량 데이터와 상관관계 분석을 실시(P-value 0.05 이하의 feature들만 선별하여 시각화 실행)
분석 결과
-
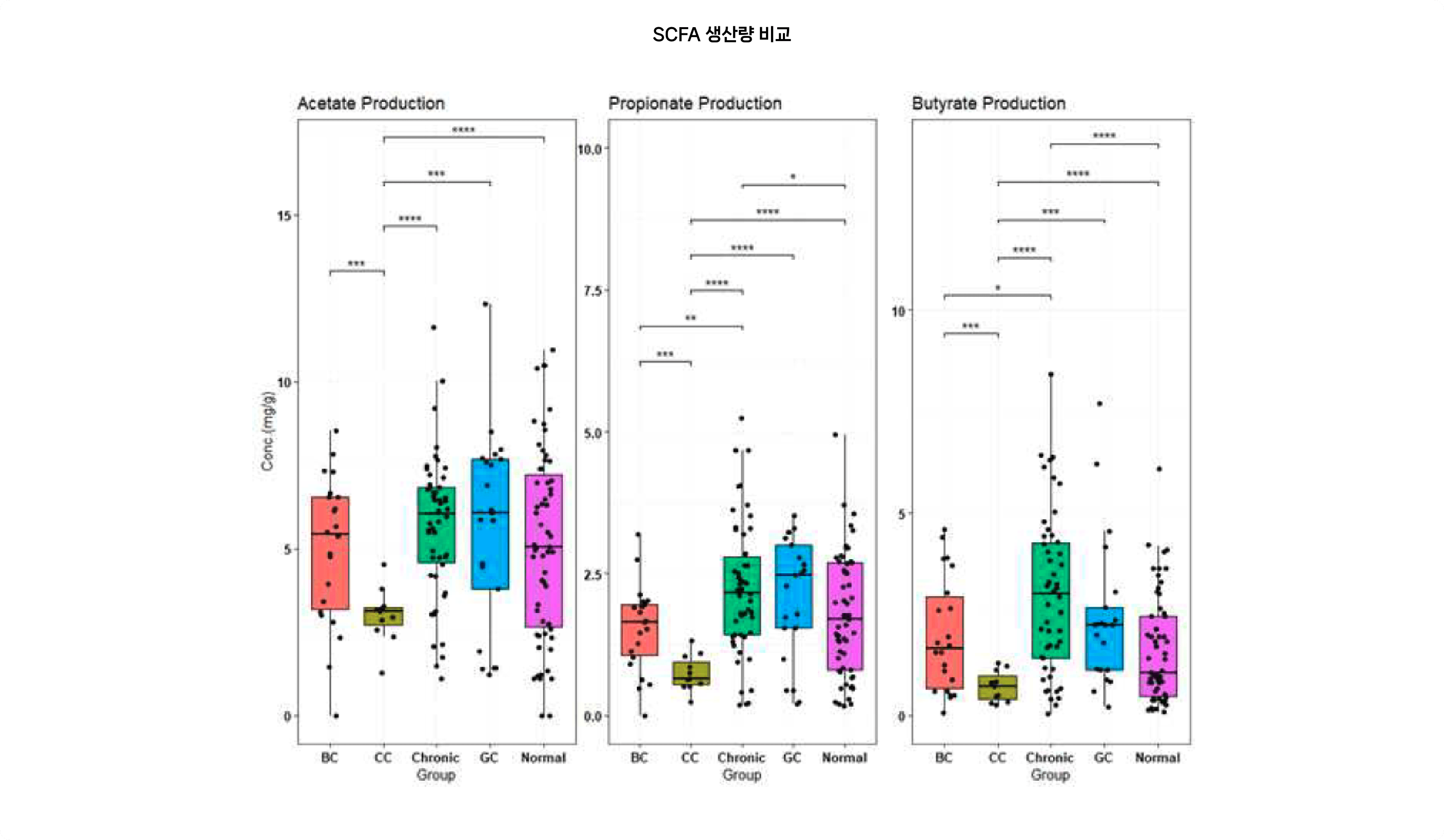
대사체(SCFA; Short Chain Fatty Acid) 생산량 비교
* 장내 생산되는 SCFA 들중 90%이상을 차지하는 Acetate, Propionate, Butyrate 생산량을 Group별로 Box plot으로 나타내었고, t.test를 통해 유의성을 검증
* 결과를 통해 볼 때, 대장암 생존자 그룹에서 3가지 SCFA 모두 현저히 낮은 생산량을 보이는 것을 확인할 수 있음.
-
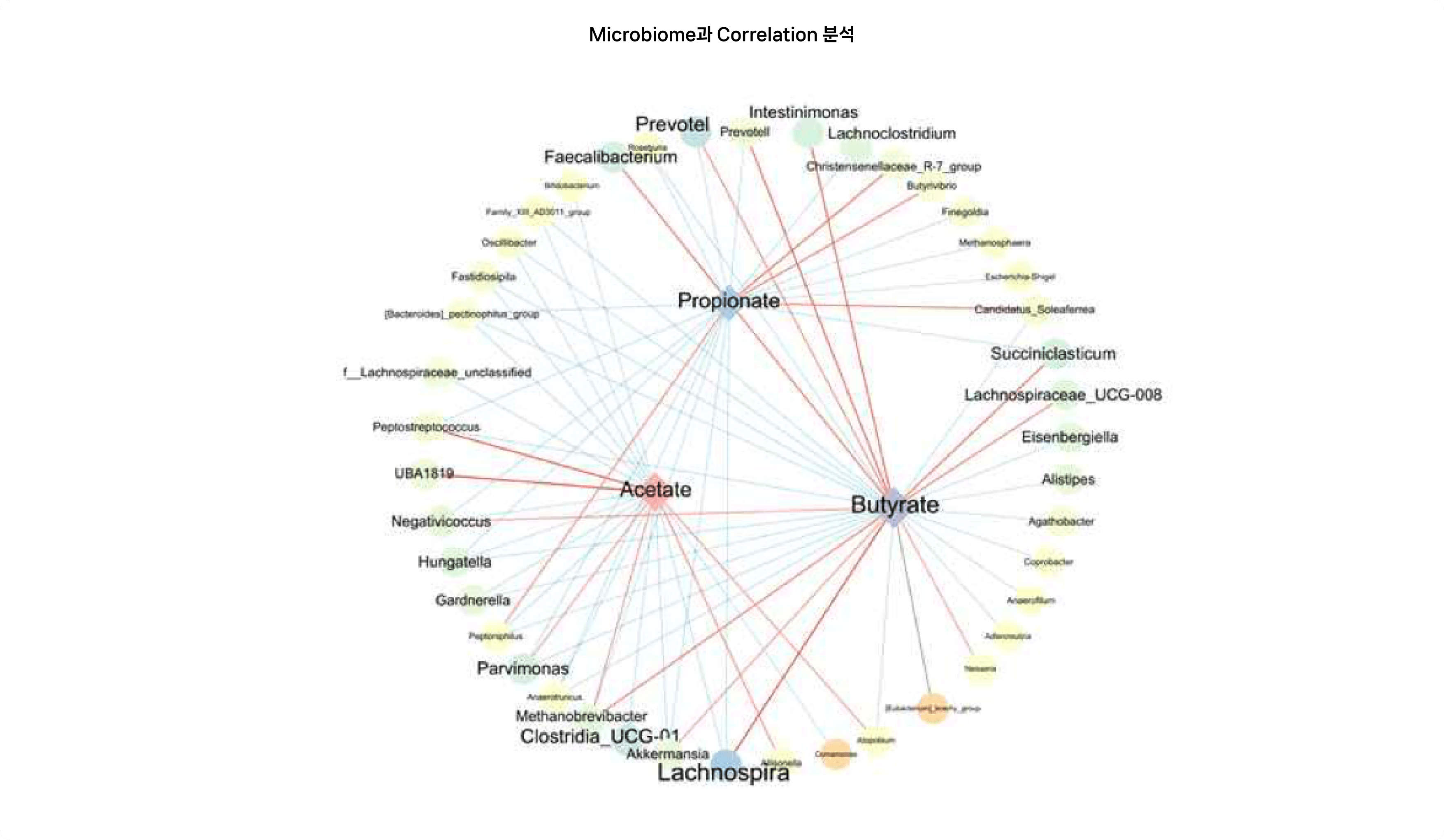
SCFA와 장내미생물의 상관관계 분석
* SCFA 생산량과 전체 샘플의 Microbiome의 상관관계분석을 진행
* 양의 상관관계를 적색으로, 음의 상관관계는 청색으로 표현하였으며 대표적으로, Butyrate의 생산과 Lachnospira가 가장 높은 양의 상관관계를 보임.
* Faecalibacterium, Prevotella 등 butyrate 생산균으로 알려진 균들 또한 Butyrate 생산량과 양의 상관관계를 보임.
-
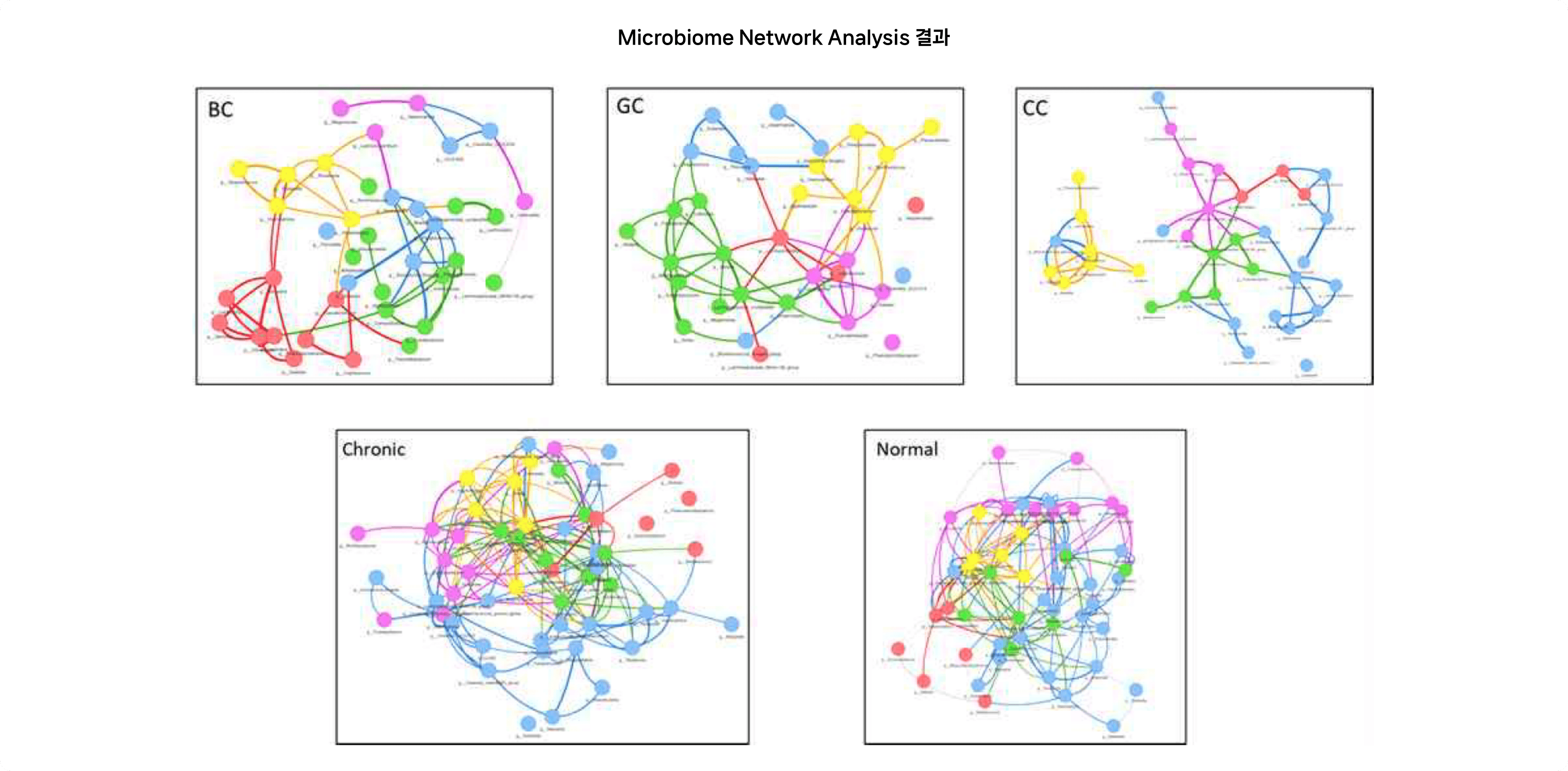
Microbiome Network Analysis (Correlation Analysis)
* 이를 통해 각 미생물 생태에서 서로의 Abundance에 영향을 주는 균들을 나타내었고, 각 Cluster는 Spearman Correlation 을 이용함.
* 만성질환자 와 일반인 그룹과 비교 했을때, 대장암을 비롯한 암 생존자 그룹에서 훨씬 단순한 Network 를 볼 수 있음.
성과 및 활용 방안
- 장내미생물에 의해 생산되는 Short Chain Fatty Acids 대사체의 생산량을 그룹간 비교함으로 대장암 생존자 그룹에서 현저하게 SCFAs 생산량이 적음을 확인함.
- 위의 분석 결과들을 통해 장내 마이크로바이옴 데이터와 SCFA 생산량의 데이터를 이용한 연관 분석이 대장암을 비롯한 인체 건강에 대한 진단 지표로 이용될 가능성을 보여줄 수 있다고 사료됨.
전장 유전체 시퀀싱(Whole Genome Sequencing) 데이터를 활용한 조직적합성항원(HLA) 유형 분석 사례
목적 및 배경
약물에 의해 유발되는 중증피부유해반응은 발생률은 낮으나 발생 시 치명도가 높은 중증 질환으로, 그 중 일부는 HLA 유전자형을 통해 예측할 수 있음.
HLA 유전자형은 인구집단 별로 빈도 분포가 상이하여 같은 HLA 유형이라도 인구집단 별 민감도 차이가 큰 특성을 보임.
- HLA 유전자형에 기반한 임상지침을 약물 부작용 예방에 보다 효과적으로 활용하기 위해서는 한국인에서의 HLA 유전자형의 빈도 분포에 대한 종합적인 파악이 필요함.
헬스케어 쇼케이스 빅데이터 분선센터에 사전 저장된 전장유전체 시퀀싱데이터를 활용하여 HLA 유전자형 추출이 가능하므로, 약물부작용의 선제적 예방을 위하여 인구집단 내 다양한 HLA 유전자형의 분포를 파악하고 안전성 확보를 위한 임상 지침 마련으로의 활용 가능성을 분석함.
활용 데이터
- 헬스케어 쇼케이스 빅데이터 분석센터에 기구축된 전장 유전체 시퀀싱 데이터(WGS)
분석 Tool
- Hisat-genotype
연구 내용
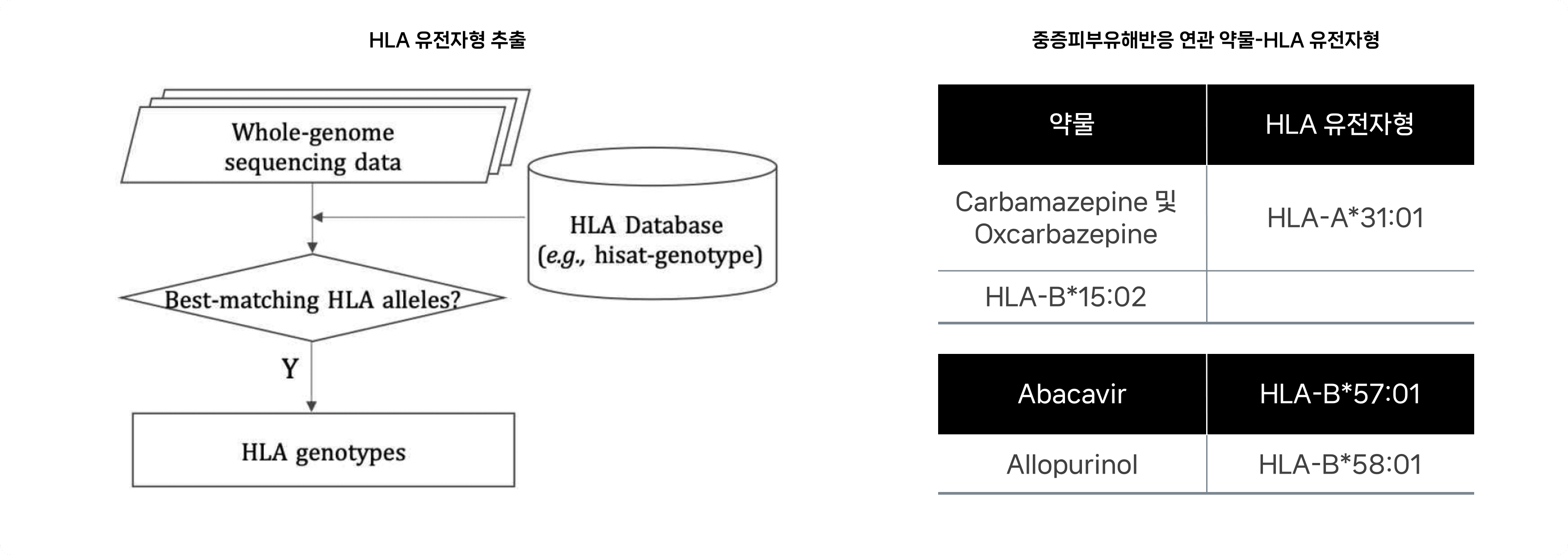
- Hisat-genotype 툴을 이용하여 사전 저장된 396명의 한국인 전장유전체 시퀀싱 데이터로부터 7개의 주요 HLA 유형에 대한 샘플 별 유전자형 획득(7개의 HLA 유형: HLA-A, B, C, DMA, DMB, DOA, DOB)
- 약물-유전형 가이드라인이 존재하는 2개의 HLA 유형 (HLA-A, B)에 대한 샘플별 유전자형 추출 (Field 2 기준)
- 중증피부유해반응 연관 약물의 주요 HLA 유전자형 (HLA-A*31:01, HLA-B*15:02, HLA-B*57:01, HLA-B*58:01)에 대한 인종별 빈도 분포 비교 표 제시
- 한국인 및 동양인 특이적 분포를 보이는 HLA-B*58:01 유전자형의 인구집단 빈도 비교 분포 지도의 확립
분석 결과
- 396명의 한국인 유전체 데이터에서 7개의 주요 HLA 유형에 대한 유전자형 분포 그래프
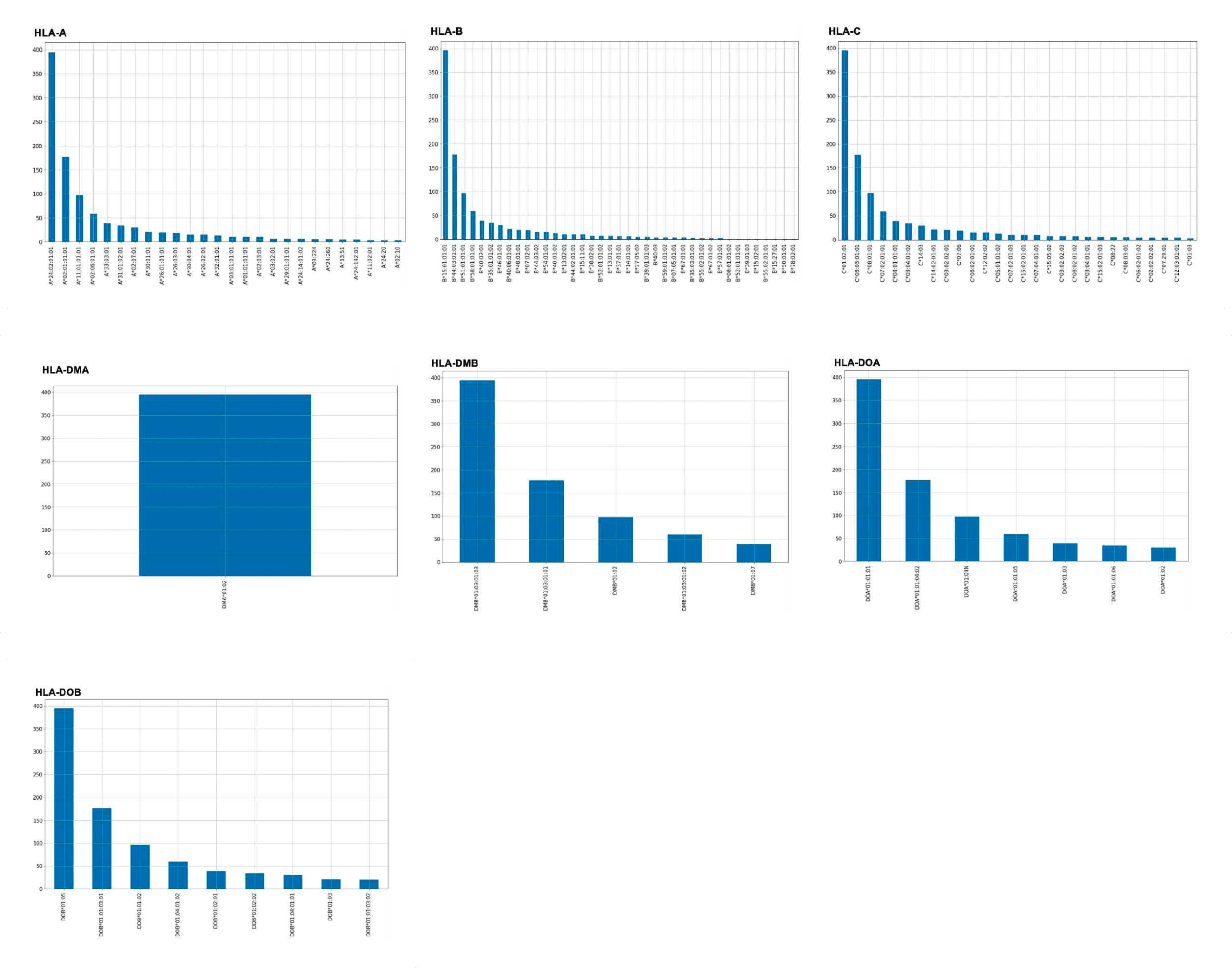
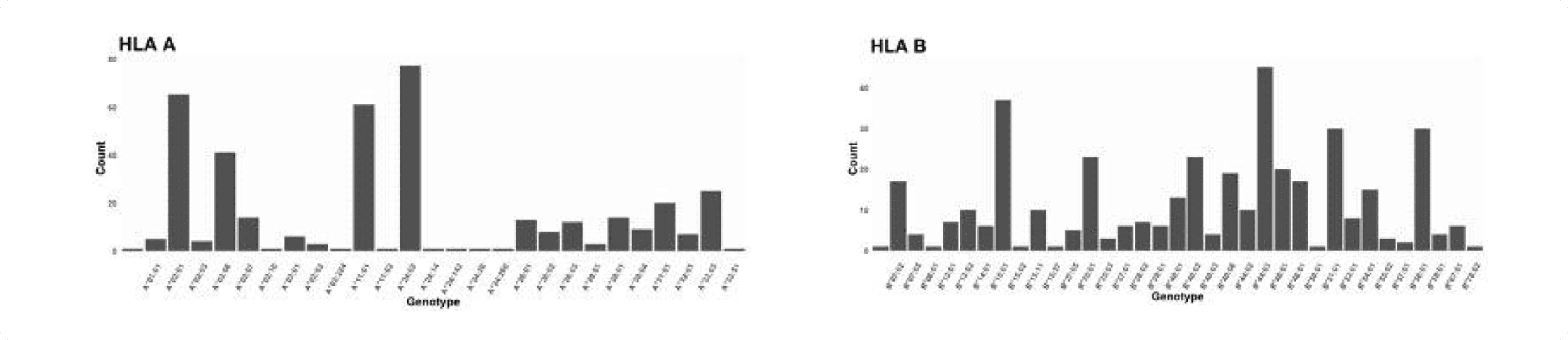
- HLA-A/B의 sub-allele 별 빈도 분포 (Field 2 기준)
- 중증피부유해반응 연관 약물의 주요 HLA 유전자형 (HLA-A*31:01, B*15:02, B*57:01, B*58:01) 에 대한 인종별 빈도 분포표

성과 및 활용 방안
- 396명의 전장유전체 시퀀싱 데이터를 활용하여 주요 HLA 유형에 대한 한국인에서의 유전자형 분포를 확인하고, 인종간 빈도 분포 비교를 통하여 한국인 및 동양인 특이적으로 발견되는 HLA 유형을 확인함.
- 특히, 약물-유전형 가이드라인이 존재하는 2개의 HLA 유형 (HLA-A, B)에 대한 샘플 별 유전자형 추출을 통하여 약물 부작용의 선제적 예방을 가능하게 하는 유전자형 기반 임상 가이드라인의 활용 가능성을 확인함.
- 중증피부유해반응 연관 약물의 네 가지 주요 HLA 유전자형 (HLA-A*31:01, HLA-B*15:02, HLA-B*57:01, HLA-B*58:01)에 대한 인종별 빈도 분포 비교를 통해 잠재적 고위험 환자군의 분포를 파악함.
- HLA-B*58:01은 한국인 및 동양인에서 높은 빈도로 발생하는 HLA 유형으로 allopurinol 유발 중증피부유해반응과 강한 연관성이 보고되어 있음. CPIC 가이드라인에 따르면, HLA-B*58:01 보유자의 경우 allopurinol을 투약하지 말 것을 권장하고 있음.
- 본 연구를 통해 얻은 유전 정보에 기반한 개인별 예상 약물반응은 보고서 등의 형식으로 개인에게 반환할 수 있으며, 이러한 맞춤형 임상지침 제공은 치료제의 안전성 확보 및 치료 효과 증진에 도움이 될 것으로 기대됨.
전장유전체 시퀀싱데이터를 활용한 유전체 기반 약물 처방 임상 지침 제공 사례
목적 및 배경
최근 의학은 치료 중심에서 예방 중심으로 그 패러다임이 변화하고 있으며, 약물유전체 데이터를 활용한 정밀의료는 예방의학의 중심에 있음.
CPIC 가이드라인은 약물-유전자 쌍에 대한 다양한 연구 결과를 바탕으로 마련된 약물 처방에 대한 임상지침으로, 임상적으로 중요도가 높은 약물-유전자 쌍(Level of evidence: A, A/B)의 경우 실제 임상 진료에 활용되고 있음.
헬스케어 쇼케이스 빅데이터 분석센터에 사전 구축된 전장유전체 시퀀싱 데이터로부터 약물 유발 부작용과 연관 있는 유전자형을 추출하고 이를 기반으로 개인에 대한 예상되는 약물반응을 유추할 수 있음.
- 즉, 약물 사용 전 예상 반응성을 제시함으로써 예방적 조치를 취할 수 있음.
유전 정보에 기반한 개인별 예상 약물반응은 보고서 등의 형식으로 개인에게 반환할 수 있으며, 개인의 유전적 다양성을 고려한 맞춤형 임상지침 제공은 치료제의 안전성 확보 및 치료 효과 증진에 도움이 될 것으로 기대됨.
활용 데이터
- 헬스케어 쇼케이스 빅데이터 분석센터에 기구축된 전장유전체 시퀀싱 데이터
분석 Tool
- Stargazer
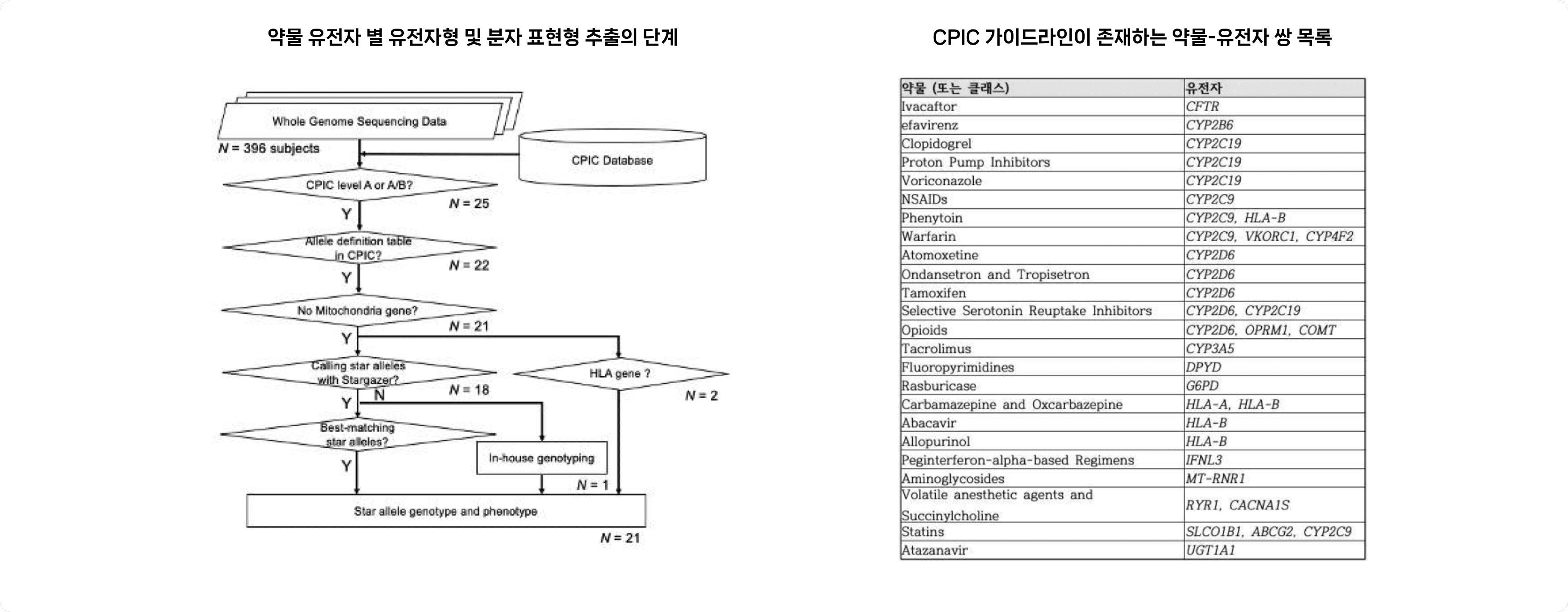
연구 내용
- 396명의 개별 환자 전장 유전체 시퀀싱 데이터로부터 약물 유전자 영역 유전체 정보를 추출
- 79개 약물 및 25개 유전자에 대한 star-allele 기반 일배체형 (haplotype) 및 유전자형 (diplotype) 추출
- 약물 유전자 별 유전자형과 매치되는 예상 분자 표현형 추출
- CPIC 가이드라인에 기반하여 분자 표현형에 따른 약물 별 권장 처방 지침 제공
분석 결과
- 약물 유전자 별 일배체형 및 예상 분자 표현형의 분포
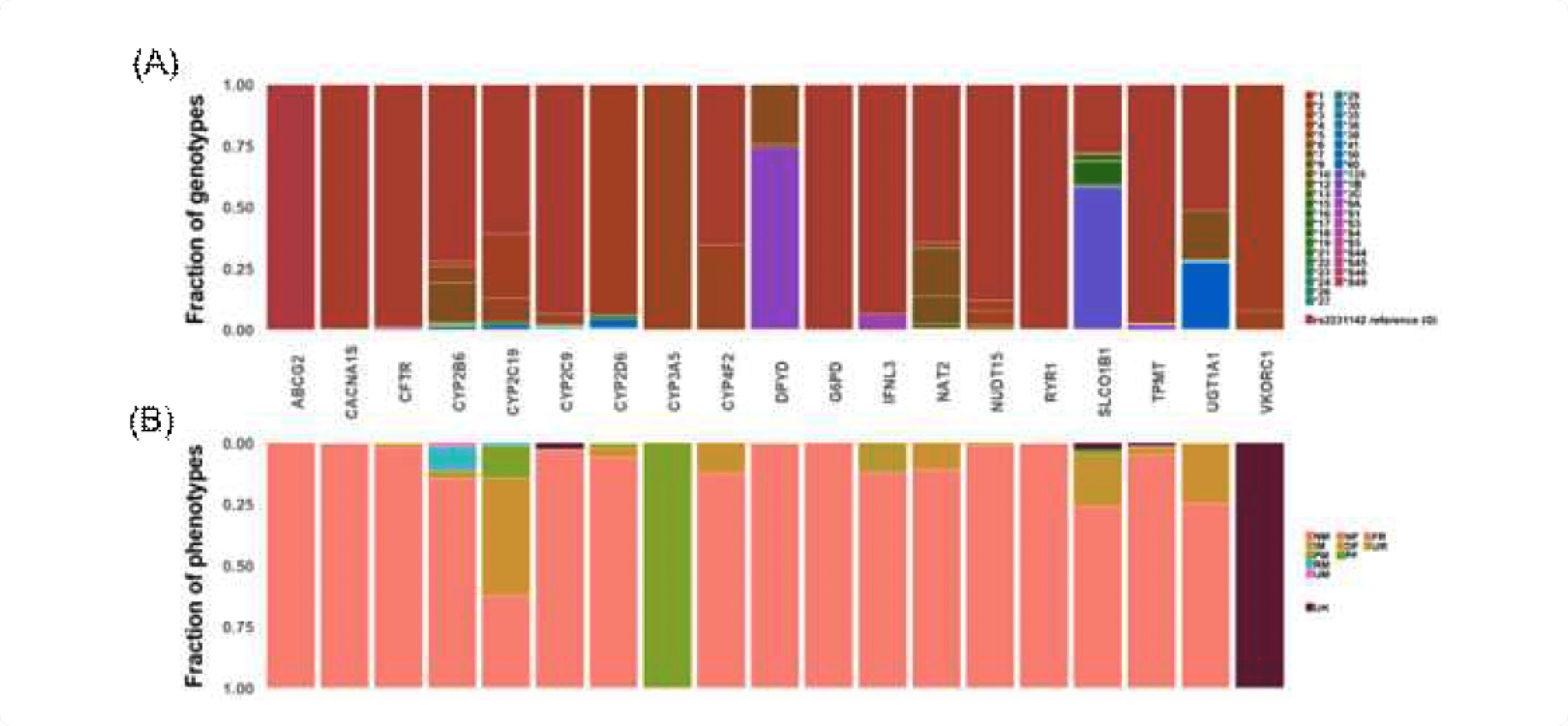
- 19개의 CPIC level A and A/B 유전자에서의 star-allele 유전자형 분포 (HLA 유전자 제외)
- 396명에 대한 유전자 별 예상 분자 표현형 분포
-
약물 유전자 별 일배체형 기반 예상 기능의 분포
* CPIC에서 일배체형-기능의 매칭 테이블을 제공하는 10개 유전자에 대한 결과 분포
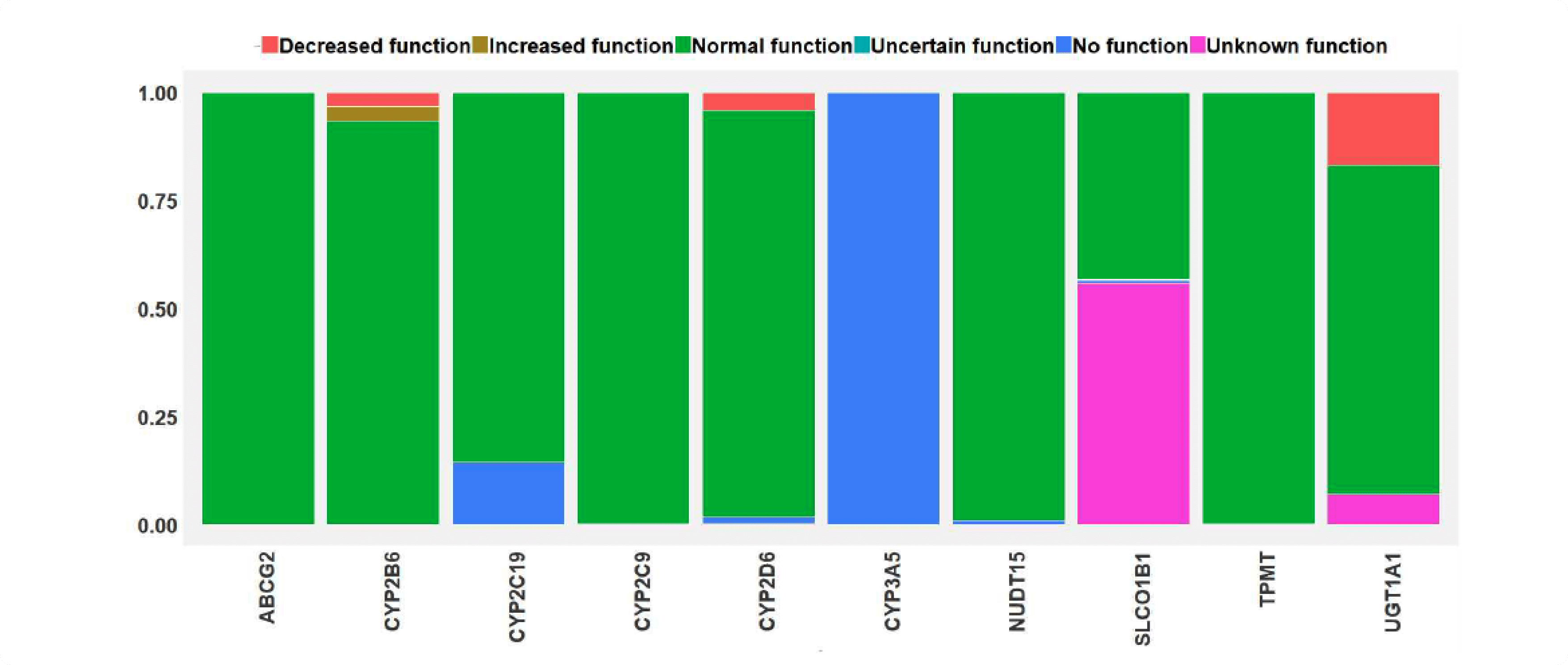
-
SLCO1B1 유전자형 기반 약물 처방 가이드라인
* (예제) Statin 계열 약물에 대한 유전자형 기반 처방 가이드라인
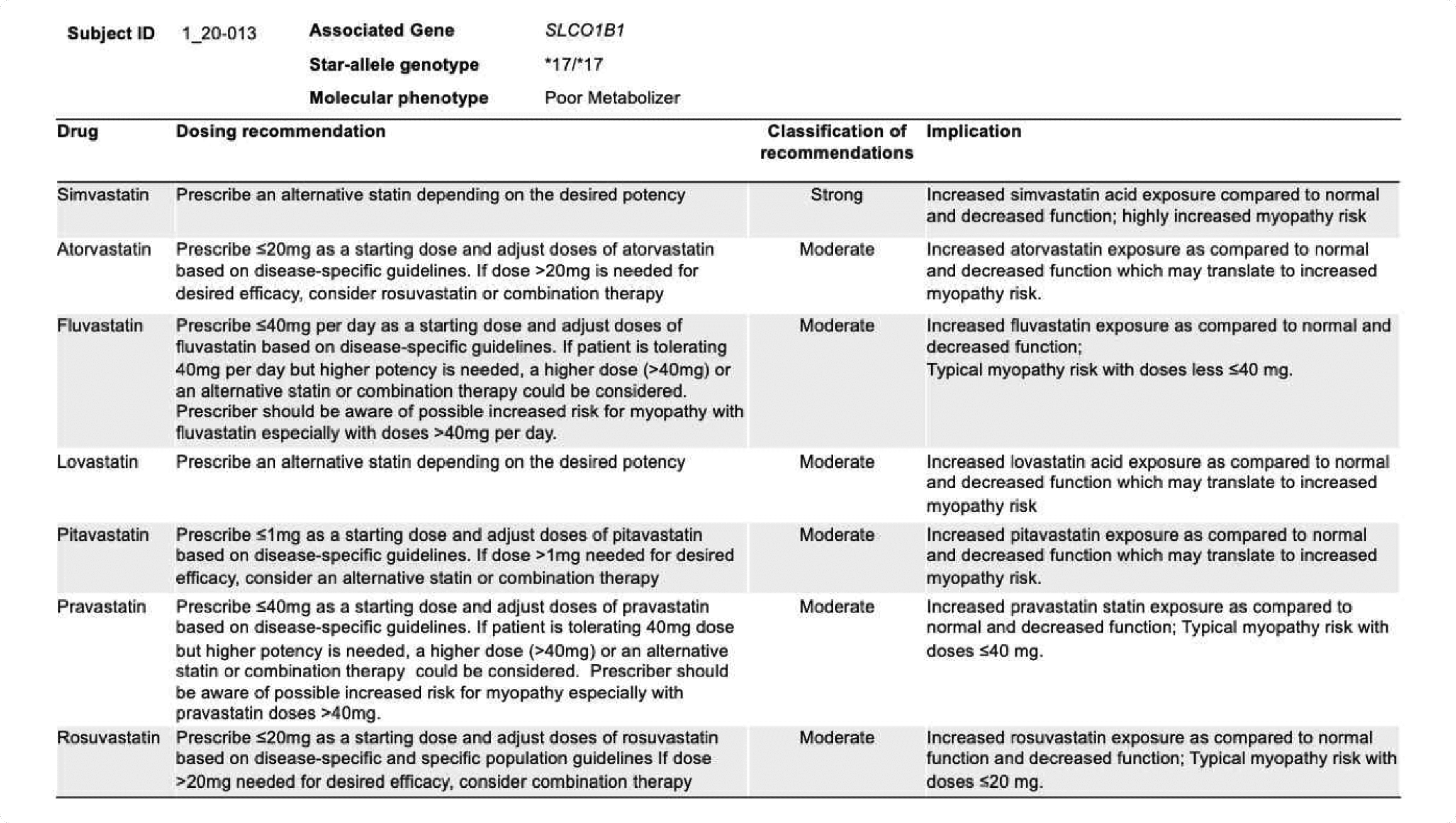
성과 및 활용 방안
- 전장유전체 시퀀싱 데이터를 활용하여 19개 주요 약물 유전자에 대한 환자별 약물 반응성 예측 결과를 얻음.
- 추출한 분자 표현형에 근거하여 CPIC 가이드라인 기반의 환자별 맞춤형 처방 지침 권고안을 제시함.
-
특히 본 연구에서 타겟으로 한 임상적으로 중요도가 높은 약물-유전자 쌍 (Level of evidence: A, A/B)의 경우, 실제 임상에서 약물 처방을 위한 임상지침 결정에 supporting evidence로 활용할 수 있음.
* 1_20-013 환자의 경우 SLCO1B1*17/*17 유전형 보유자로 statin 계열 약물의 대사 능력이 현저히 저하되어 있는 것으로 예측 되며 CPIC 가이드라인에서는 특히 simvastatin 약물 복용시 근질환 (myopathy)의 위험이 높으므로 대체 약물을 사용할 것을 강력하게 권장하고 있음.
- 유전정보에 기반한 개인별 약물 반응 예측 결과는 보고서 등의 형태로 개별 환자에게 반환될 수 있음.
- 임상에서는 선제적 유전체 검사를 통해 예측 가능한 약물유발 부작용 예방에 적극 활용(약물의 점진적 증량을 통한 tolerance 유도 및 대체약제 투여 권장)할 수 있음.